
This website uses cookies to improve your experience. We'll assume you're ok with this, but you can opt-out if you wish. Privacy & Cookies policy
A new state-of-the-art 3D Quantitative Structure-Activity Relationship (QSAR) software package that builds statistical models (CoMFA, CoMSIA and HyPhar) based on data obtained from experimental assays.
Many molecular properties can be predicted based on available information extracted from chemical structures and experimental assays. However, most of the available and used methods neglect the 3D geometrical properties of the molecules, which play a critical role in understanding the interaction between a ligand and a receptor. These properties are even more important when considering bioactive conformations of the molecules derived from experiments.



We developed a 3D Quantitative Structure-Activity Relationship (QSAR) software package that builds statistical models (CoMFA, CoMSIA and HyPhar) based on data obtained from experimental assays.
Our tool uses a unique and superior 3D representation of molecules based on electrostatic, steric and hydrophobic interaction fields derived from semi-empirical Quantum-Mechanics (QM) calculations. Such fields describe with high accuracy the factors that determine ligand / receptor interactions. PharmQSAR is peer-reviewed and validated. See our publications for more information.



Features
Applications
• Predicting molecular properties.
• Pharmacophore generation.
• Visualizing relevant areas for ligand-receptor interaction. in order to:
• Improve your candidate molecules in the Lead
Optimization phase.
• See which areas of the molecule should be modified
• Prioritize follow-up compounds based on predicted
activity.
• Predict other key molecular properties of new
compounds.
• Understand which factors drive the activity of
your leads.
• Improve your virtual screening searches in ligand libraries.
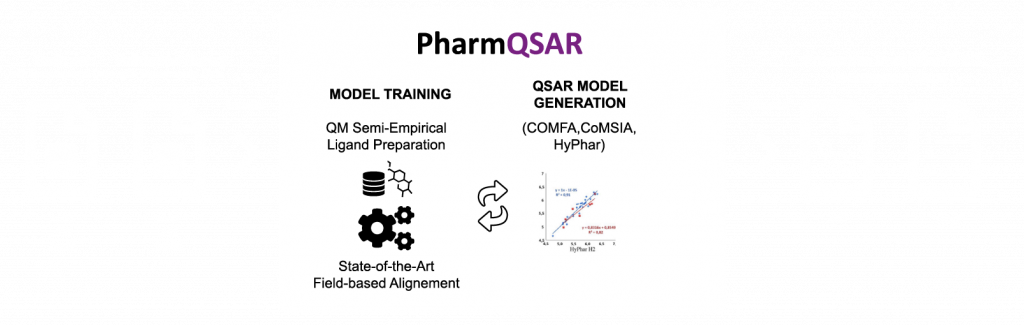
PharmQSAR allows to automatically generate 3D QSAR/QSPR models using a library of compounds with known activities or properties and their molecular fields. The generated model is validated with an external data set of molecules. The trained model permits to predict relevant properties such as important ligand-receptor interactions and evaluate these properties on new chemical libraries. PharmQSAR generates also projections for an easy visualization of the calculated properties.
Choose what works best for you and start using PharmQSAR: the versatility of the command-line interface, or the connectivity and scalability of our API with other platforms and operating systems.
Run on your IT infrastructure
Scalable calculations
Updates and support included
Seamless integration and scalability
Python library interface
AWS integration

This website uses cookies to improve your experience. We'll assume you're ok with this, but you can opt-out if you wish. Privacy & Cookies policy