By Nicola Scafuri and Ana Caballero
A larger number of ligands in the virtual screening library increases the chances of identifying ligands that are more potent, selective, or possess improved physicochemical properties. Mining such chemical spaces using the 3D representation of molecules has shown to be a successful approach, however, a reliable screening is only feasible when the 3D library is properly prepared. Getting ready a 3D library from a 2D representation is not trivial, since several chemical aspects must be considered, for example:
- A single 2D molecule can exist in multiple protomeric and tautomeric forms at a given pH, each with a distinct distribution.
- Molecules with chiral centers can exist in various 3D stereoisomeric forms.
- All potential 3D conformers must be thoroughly considered.
PharmScreen®, our field-based virtual screening software, has exhibited a promising performance in identifying novel hits within 3D libraries with similar physic-chemical properties to reference compounds. Thanks to a unique and superior 3D representation of molecules based on electrostatic, steric, and hydrophobic interaction fields derived from semi-empirical Quantum-Mechanics (QM) calculations, the internal and external benchmarks have identified a promising number of novel and diverse hits for several targets. Notwithstanding, even these encouraging results rely on the adequacy of using a 3D accurately prepared library.
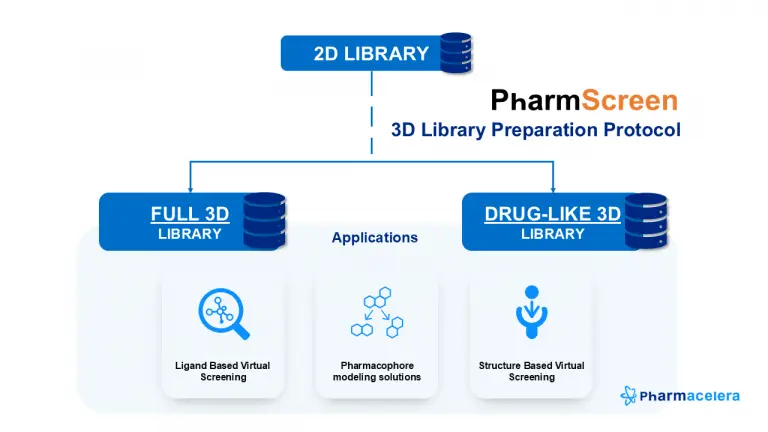
We have developed an internal protocol for preparing 3D libraries suited for ligand-based virtual screening (LBVS) and structure-based virtual screening (SBVS), such as docking campaigns, and pharmacophore modeling solutions. It aims to establish a standardized and easily reproducible protocol for preparing 3D libraries.
The protocol integrates internal scripts and the PharmScreen® software, and it was initially utilized to prepare the Enamine Screening Collection library. This library is one of the world’s largest screening compound libraries, boasting over 4.4 million unique compounds. The protocol involved the generation of different protomers and tautomers at pH 7.4, and all possible stereoisomers and conformers. As a result, our protocol ensures an extensive, high-quality chemical space, to deliver unique and tailored drug discovery solutions. Indeed, we have now one of the largest and most up-to-date 3D screening libraries of synthesizable compounds for early drug discovery projects, featuring:
- Up to 270 million conformers for LBVS
- Up to 7.9 million 3D stereoisomers for docking campaigns, optimized for pharmacophore modeling solutions
- In addition, a complete screening of this library can be achieved in just 25 hours using PharmScreen®
Our protocol can also filter the prepared 3D library based on drug-like properties to focus the hit ID towards the drug-like space, enhancing efficiency in the execution of CADD projects.
This robust protocol has shown to be extensible to other commercial libraries, for example, the Molport Screening Compounds library, further expanding the chemical space from which we can extract novel hits.