By Carlos Cruz and Ana Caballero
The success of a ligand-based virtual screening campaign relies on their molecular descriptors, in this sense, Quantum Mechanics (QM) offer higher accuracy by capturing detailed electronic properties, the influence of 3D conformation, and key interactions that classical descriptors often overlook. Pharmacelera´s property molecular descriptors exploits self-consistent reaction fields methods to define hydrophobic topologies from atomic contributions.
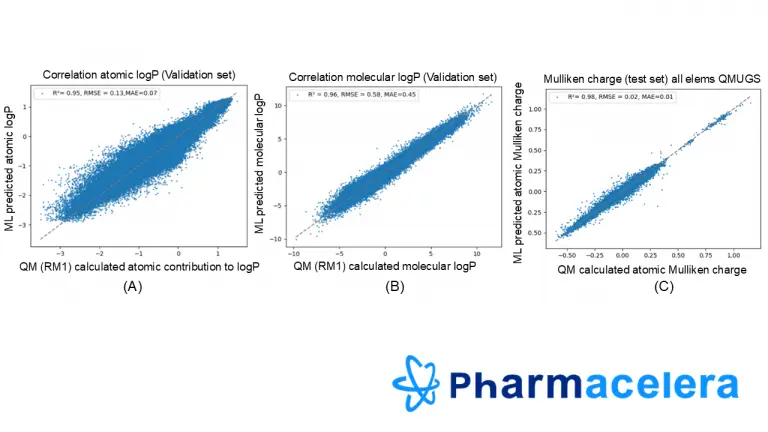
Our unique descriptors are calculated using accurate QM methods, through the Recife Model 1 (RM1) parametrization of the Miertus Scrocco Tomassi (MST) model, which is known to be a reference method due to its good balance between accuracy and calculation time. However, QM calculations are still time-consuming, especially for huge chemical spaces. To handle this challenge, our team has developed a Machine Learning model for predicting atomic logP, to determine if exploring huge chemical spaces in a reduced amount of time is possible.
Our model includes physical descriptors (ex. topological, steric, and electrostatic descriptors), transferring information about the 3D environment of the atom to the model. The precise description of each atom gave us the possibility to develop and validate a model able to predict 3D atomic contributions to logP values with an R^2 >0.9 for any kind of neutral drug-like molecule 2.000 times faster than the calculations using the RM1 parametrization (see graphics (A) and (B))
Using this atomic description, we have accurately extended it to predict other atomic properties. For example, graph (C) shows the excellent results obtained for Mulliken Charges derived from Density Functional Theory calculations from the QMUGs library.
Interested in the application of QM methods to drug discovery? Pharmacelera software uses a unique 3D representation of molecules based on electrostatic, steric and hydrophobic interaction fields derived from semi-empirical QM calculations. Discover PharmScreen, exaScreen and PharmQSAR.
Need a customized solution for your drug discovery project? Contact our team to arrange a call and discuss your current challenges.