
By Alessandro Deplano – Sep. 20, 2017
NOTE: THERE IS A NEW VERSION OF THIS SCRIPT.
VISIT THIS PAGE TO DOWNLOAD IT.
Conformer generation is one of the first and most important steps in most ligand based experiments, particularly when the ligand’s 3D structure is unknown. For example, the quality of the conformers could affect the results of virtual screening experiments.
At Pharmacelera we have written a python script to generate conformations with RDKit1, one of the best freely available tools for conformer generation due to its accuracy reproducing experimentally determined structures and its reasonable computing requirements2.
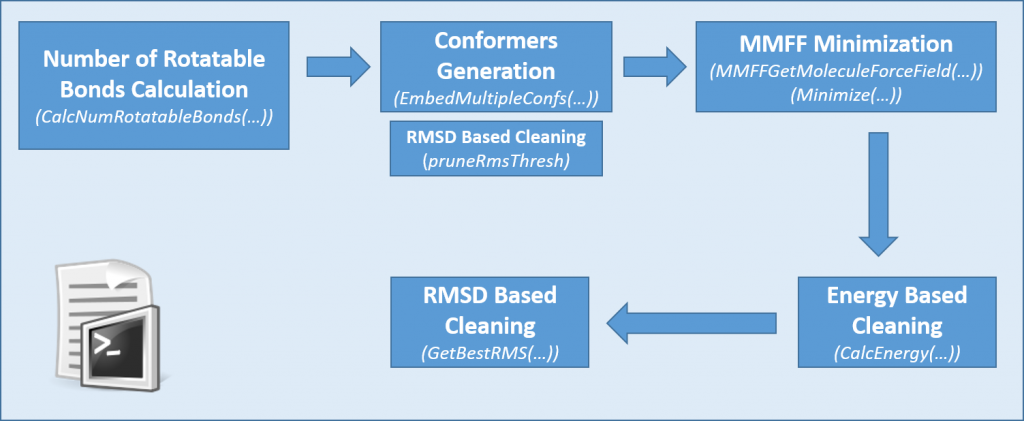
The script (Figure 1) uses RDKit functions like EmbedMultipleConfs3 and allows the generation of high quality conformers. With the usage of multiple filters this script finds the same amount of bioactive conformations than the default function but with a 57% reduction in the number of conformers.
The optimal number of conformations will vary based on molecular flexibility. It is known that the number of rotatable bonds is highly related with the size of the conformational space. Therefore, with this script users can generate either a fixed number of conformers or generate them based on the number of molecular rotatable bonds.
To establish the best relationship between rotatable bonds and the minimum number of conformers needed to find the experimentally determined structure we performed an extensive study using an AstraZeneca dataset4 composed by 1456 molecules with a spectrum of rotatable bonds from 0 to 13 (Figure 2).
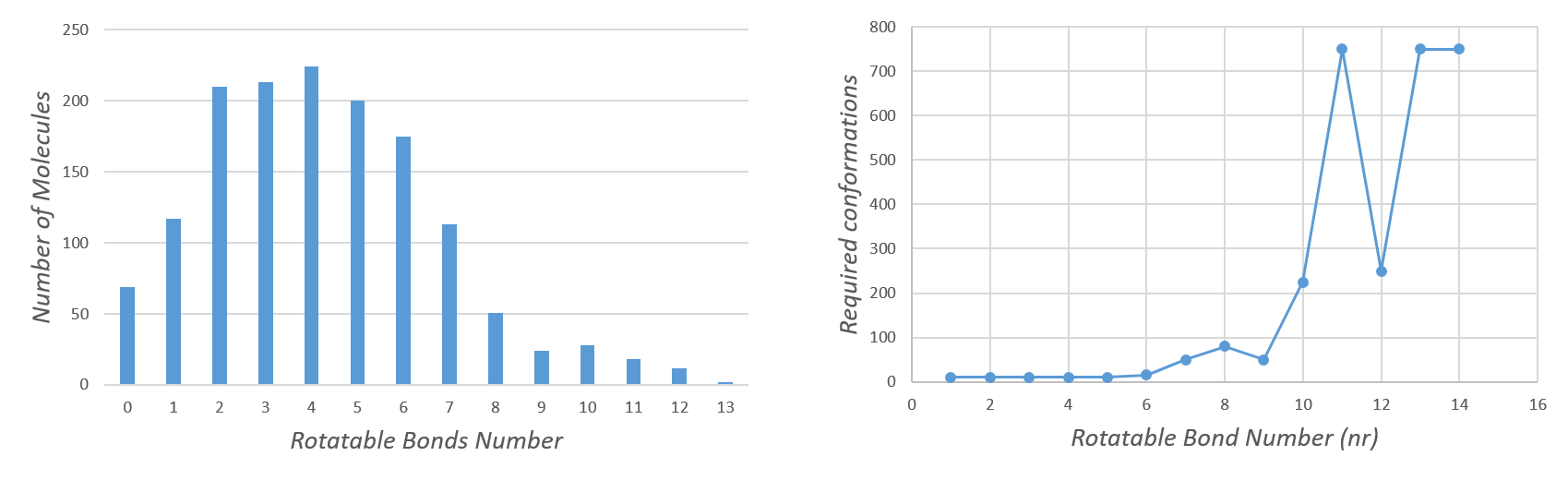
Figure 2 highlights an exponential correlation between the number of conformers and the rotatable bond to find the crystal structure. Based on these results the number of conformers for each molecule is determined by this equation:
Conformation_number = Number_of_rotable_bonds^3
Molecular energy is another important aspect in conformer generation. In fact, molecules can exist only in some range of energy. This script allows the user to set a maximum value of energy that all molecules can differ from the one with the lowest energy. In this context we have evaluated different energy windows to find the best energy threshold that minimizes the number of conformers without losing in accuracy.
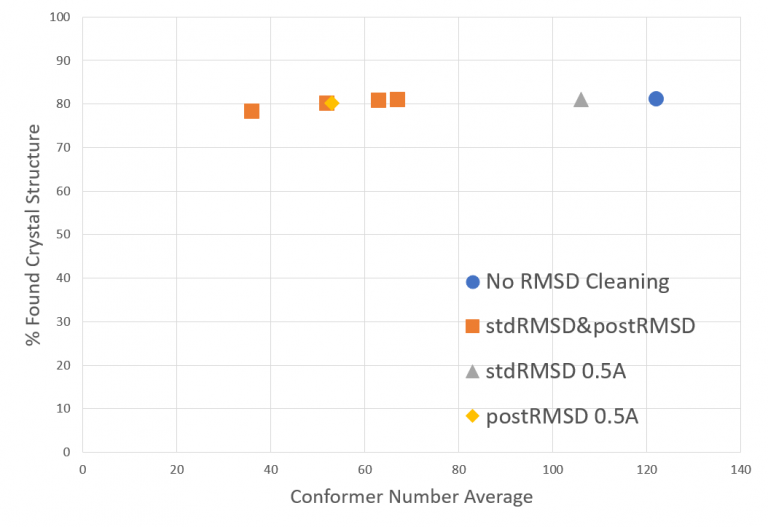
Finally, one of the most important properties of a conformer set is a proper balance between the maximum space exploration and the minimum number of conformers. Based on this assumption this script allows a RMSD-based cleaning that keeps only those conformers which differ from the others. In particular, the script performs two RMSD-based cleanings which can be used in combination or as single. The first RMSD cleaning is performed with the pruneRmsThresh option of the RDKit EmbedMultipleConfs function, which performs the cleaning before molecular minimization.Performing an RMSD cleaning before energy minimization, however, might cause that different conformers after the minimization fall into the same local energy minimum and become structurally very similar. To avoid this problem another RMSD cleaning function was added to the script which performs the purge after the minimization. Also, in this case, some experiments were performed to find the optimal RMSD cutoff value to reduce the number of conformers without influencing the accuracy of the results (Figure 3).
| Strategy | Conformer Number Average | % Crystal Structures Found |
| Default function | 122 | 81.18 % |
| Energy Cleaning 6.0 Kcal/mol | 121 | 81.18 % |
| RMSD Cleaning 0.50 Å | 52 | 80.15 % |
| Final Configuration: Energy Cleaning 6.0 Kcal/mol & RMSD Cleaning 0.50 Å | 52 | 80.01 % |
Table shows the amount of conformers generated with each script configuration option and the percentage crystal structures found. It can be seen that a post minimization RMSD cleaning is very useful reducing the required number of conformations. On the other hand, the energy cleaning does not show a significant impact in conformer reduction. However, it is kept in order to remove outliers with unreasonable energy levels.
- http://www.rdkit.org/
- Ebejer JP, Morris GM, Deane CM; (2012) Freely Available Conformer Generation Methods: How Good Are They? J Chem Inf Model 52:1146-1158.
- http://www.rdkit.org/Python_Docs/rdkit.Chem.rdDistGeom-module.html#EmbedMultipleConfs
- Giangreco I, Cosgrove DA, Packer MJ (2013) An extensive and diverse set of molecular overlays for the validation of pharmacophore programs. J Chem Inf Model 53:852–866.