

Minyi Su
May 18th, 2023
Ligand-based virtual screening is essential in modern drug discovery workflows and PharmScreen® has been shown to find novel molecules through its unique 3D representation of molecules. However, traditional high-throughput virtual screening tools have some limitations when dealing with ultra-large virtual libraries in terms of computational cost and time. To address this issue, multiple techniques have appeared that use machine learning methods to handle these libraries. Among them, the MolPAL protocol developed by Coley et al. [1] is a promising alternative to solve this problem. Pharmacelera has used the MolPAL protocol to accelerate PharmScreen® simulation on large libraries and validated it on two GPCR targets: Histamine H1 and Muscarinic 4 (M4).
MolPAL employs a Batched Bayesian Optimization strategy for sampling promising compounds among the library (Figure 1). First, a small subset of the library is randomly selected and evaluated using PharmScreen®. A surrogate machine-learning (ML) model is then trained on this subset and used to predict acquisition scores for the complete library. The top N scored compounds that were not selected in the previous iteration are then selected to run the PharmScreen® simulation. These steps are run iteratively until a stopping criterion is met. Users can observe promising compounds with top similarity scores among all selections with a significant reduction in computational cost. Compared to exhaustive sampling in brute force simulation, only ~2.4% of the library is selected to run PharmScreen® simulation in the MolPAL protocol.
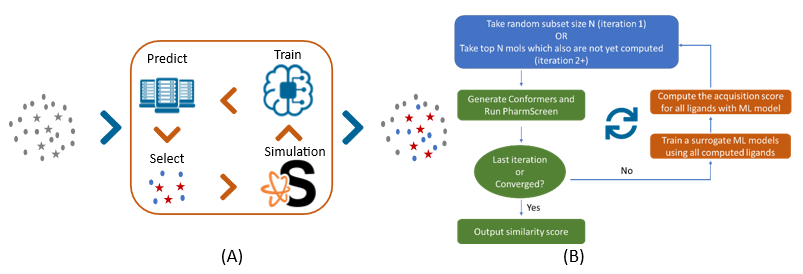
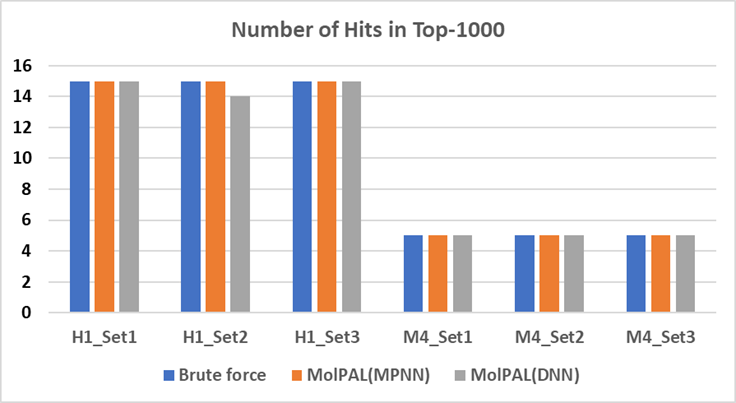
In order to validate the suitability of this protocol, the MolPAL protocol and PharmScreen® have been used with two GPCR targets: Histamine H1 and Muscarinic 4 (M4). For each target, three different libraries (1M) were built in-house and known hits were also added to the library. Both brute force and MolPAL active learning methodology were validated, and Deep Neural Network (DNN) and Message Passing Neural Network (MPNN) were also tested in the surrogate ML model construction. The histograms in Figure 2 are the number of known hits detected in Top-1000 scored compounds and results showed that the MolPAL protocol could not only speed up the simulation but also find the same number of hits as it is in brute force. This methodology can be employed to screen ultra-large libraries in drug discovery projects.
[1] Graff DE, Shakhnovich EI, Coley CW. Accelerating high-throughput virtual screening through molecular pool-based active learning. Chem Sci. 2021 Apr 29;12(22):7866-7881. doi: 10.1039/d0sc06805e.
[2] Coleygroup. (n.d.). GitHub – coleygroup/molpal: active learning for accelerated high-throughput virtual screening. GitHub. https://github.com/coleygroup/molpal/tree/main