Today, the amount of data generated in many fields such as engineering, social sciences or medicine is suffering a tremendous scale-up. Extraction of relevant information is hence becoming increasingly challenging, and methods of data analysis such as clustering are now crucial. Clustering consists on the identification of homogeneous subgroups among a set of heterogeneous items. In the context of computer-aided drug discovery, where the chemical space is estimated to be 1063 compounds, we can use it to select promising subgroups inside a large chemical library, getting rid of the bulk of the dataset with either no medical interest or redundancy a priori.
Non-hierarchical methods
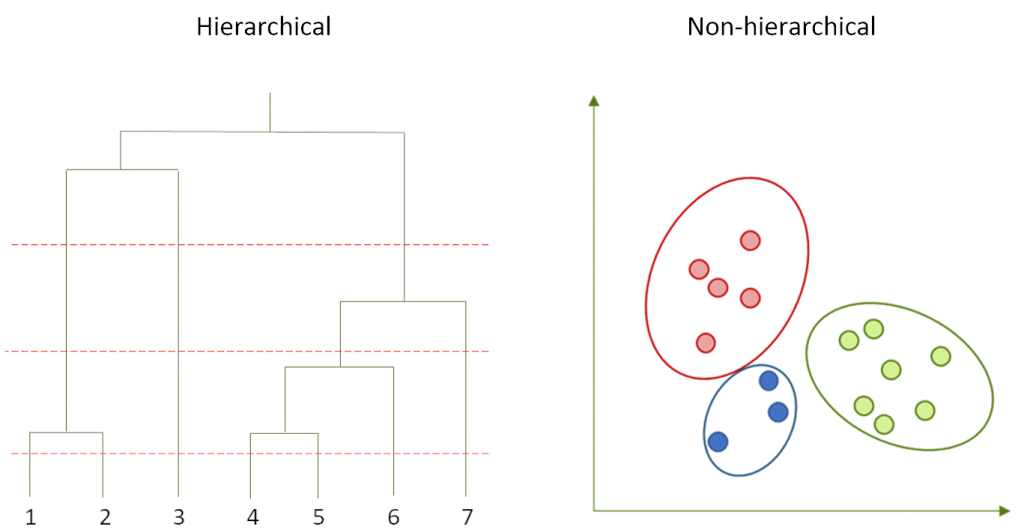
Clustering algorithms vary largely on their efficiency, so some methods are more practical than others for their application on libraries of different size (Downs & Barnard, 2002). For large datasets, the most suitable are non-hierarchical algorithms, that are based in a single partition of the data. From them, probably the most widely used is K-means (Forgy, 1965), that efficiently generates a user-defined number of clusters that are iteratively updated until an optimal classification is found. Compared with the family of nearest-neighbor algorithms such as the one developed by Darko Butina (Butina, 1999), K-means produces groups with a homogeneous size, although this sometimes comes at a cost of a higher internal heterogeneity (Walters, 2019). A faster implementation of K-means, called Mini Batch K-means (Sculley, 2010) uses just a small portion of the data to update the clusters in each iteration.
Our approach
To assess the utility of Mini Batch K-means, we generated a dataset by combining all sets of the Directory of Useful Decoys with the library from Specs, considered as decoys. In total, the dataset contained more than 300K structures from which just 2,2K were active compounds. Applying our proposed multistep protocol that combines clustering with 3D hydrophobic fields overlays, we were able retrieve a significant number of hits, screening less than a 5% of all compounds, and more importantly with high chemical diversity.
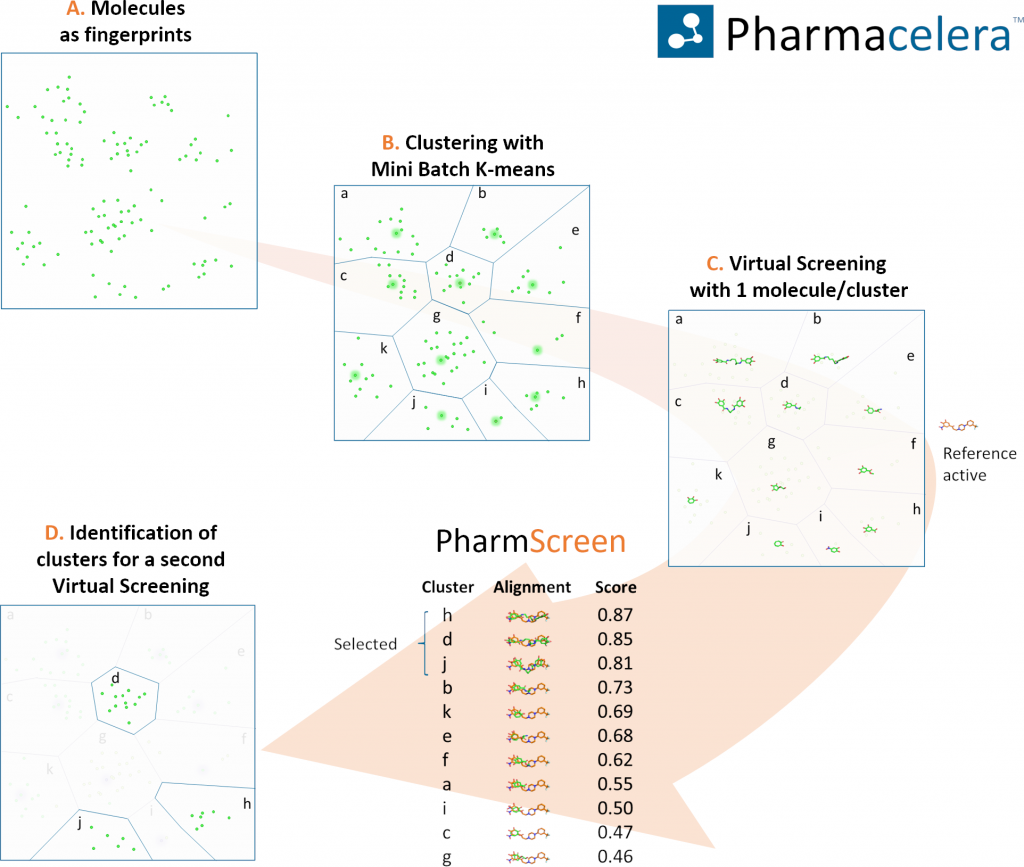
Although applying clustering methods might hide interesting structures compared to the integral screening of all compounds in a dataset, it also enables exploring a much larger chemical space that, otherwise, would be intractable. Future steps should be focused on finding the optimal number of clusters to generate, as well as increasing the quality of the chosen representative molecules.
Which is your experience with clustering large chemical libraries? How do you think we should cope with these large datasets?
- Butina, D. (1999). Unsupervised data base clustering based on daylight’s fingerprint and Tanimoto similarity: A fast and automated way to cluster small and large data sets. Journal of Chemical Information and Computer Sciences, 39(4), 747–750. https://doi.org/10.1021/ci9803381
- Downs, G. M., & Barnard, J. M. (2002). Clustering Methods and Their Uses in Computational Chemistry. In Reviews in Computational Chemistry, Volume 18 (pp. 1–40). Hoboken, New Jersey, USA: John Wiley & Sons, Inc. https://doi.org/10.1002/0471433519.ch1
- Forgy, E. (1965). Cluster analysis of multivariate data : efficiency versus interpretability of classifications. Biometrics, 21, 768–769.
- Sculley, D. (2010). Web-Scale K-Means Clustering. Proceedings of the 19th International Conference on World Wide Web, 1177–1178.
- Walters, W. P. (2019). K-means Clustering. Retrieved from http://practicalcheminformatics.blogspot.com/2019/01/k-means-clustering.html