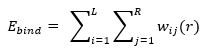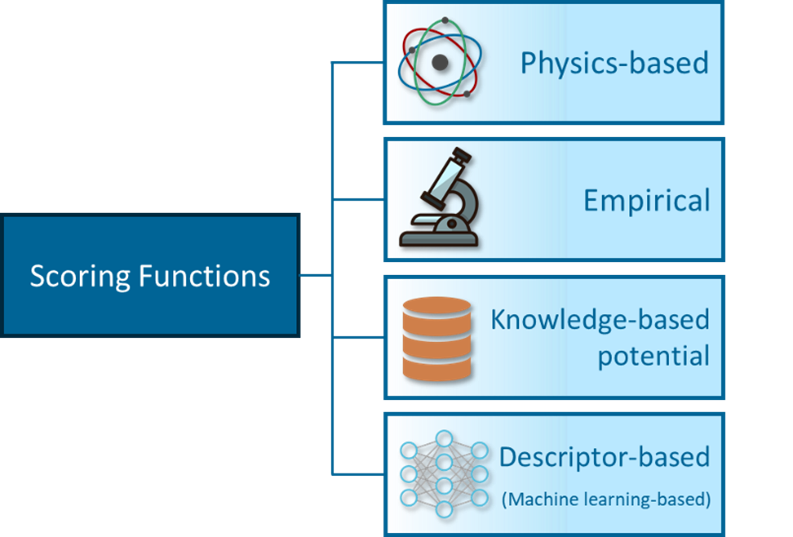
Physics-based SFs were the first ones to be developed and they are based on the physical atomic interactions between target and ligand. These are usually divided in Van der Waals forces, computed as Lennard-Jones potentials, and electrostatic forces, calculated using Coulomb’s Law. Since this approximation neglects both entropic and desolvation contributions, very often physics-based SFs incorporate additional terms to account for them. Examples of this kind of SFs are GoldScore in the docking software GOLD and SFs in Autodock 3 and 4.
![]()
Empirical SFs are probably the most intuitive approach among the four groups. They compute binding affinity by adding up a set of weighted energy terms. These terms represent energetic factors for the binding, such as hydrophobic effects, hydrogen bonds, halogen bonds, steric clashes, etc. Multiple Linear Regression is used to infere the weights from a training set of protein-ligand complexes with known binding affinities. Again, some of these terms are often devoted to account for the entropic and desolvation effects. Examples of empirical SFs are ChemScore from GOLD and GlideScore SP in Schrödinger’s Glide.
![]()
Knowledge-based SFs are based on the inverse Boltzmann statistic principle. This principle assumes that, in a training set of protein-ligand complexes, the frequency of a pair of atoms at a specific distance is directly proportional to the contribution of this pair to the binding energy. Hence, frequencies of different pairs are converted into distance-dependent potentials, and the binding affinity can be then computed as the sum of all pairwise potentials present in the complex.