We have been working hard to bring up a new Graphical User Interface (GUI) based on feedback from many computational and medicinal chemists using PharmScreen. Today we are glad to announce that the version 2.0 of PharmScreen GUI has been released.
Apart from different minor changes, the major improvements are related to ligand/library preparation and the output files in order to make PharmScreen easier to use.
Support for ligand/library preparation
The preparation of all the molecules for a correct 3D molecular modeling setup is a key preliminary step when performing a virtual screening experiment. It involves the calculation of the parameters and conformations for each molecule on the database. This step is now completely integrated in PharmScreen!
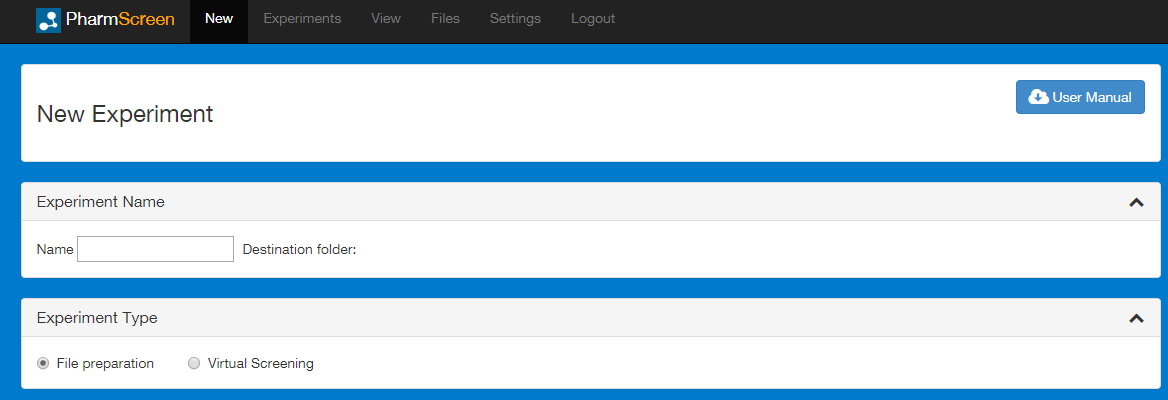
In the Experiment Type area, you can choose whether you want to prepare the library or directly launch a virtual screening experiment. Selecting File preparation, a library of molecules and the reference structures can be prepared for a PharmScreen virtual screening experiment.

For ligand preparation, you can directly upload your molecule file or work with one already loaded on the cloud platform. In this GUI version support for new input files such as SMILES and InChI has been included.
Ligands can be prepared using the new PharmScreen features like:
- Structure minimization, with two different forcefields supported (AM1 and RM1)
- Conformation generation, performed with a distance geometry algorithm that generates a variable number of conformers based on the amount of rotatable bonds (Learn more about our conformer generation process)
For each molecule (library and ligand), different parameter calculations are included:
- Partial charges can be computed using Gesteiger, Mulliken or Electrostatic
- Atomic-level LogP partitioning with semi-empirical (RM1) IEF/PCM-MST solvation models
More detailed output files
In this new GUI version, the output files of a virtual screening experiment with PharmScreen have more details:
- Output files include the Similarity Index, the SMILES and InChi codes of the structure and the reference structure for which the similarity index is printed
- The energy of each conformation is printed in the output file if conformers are generated by the GUI
Furthermore, small changes and fixes has been applied to the whole interface to make it more usable.
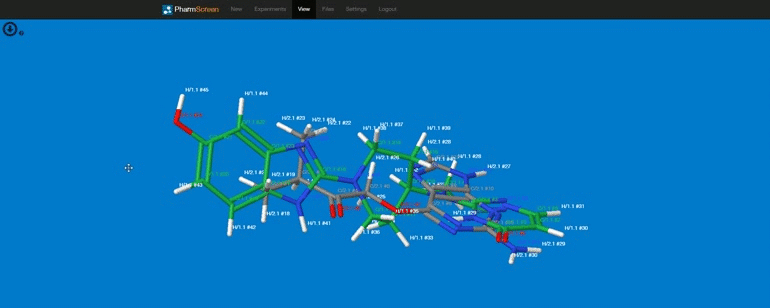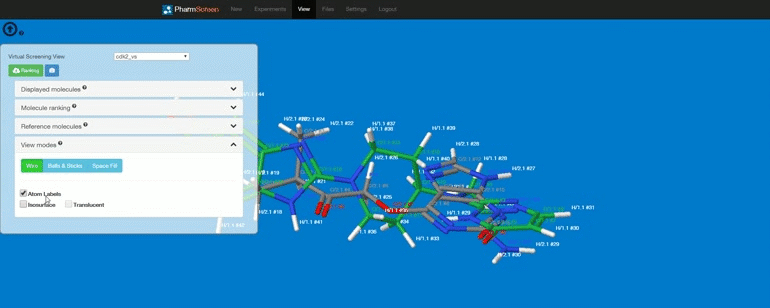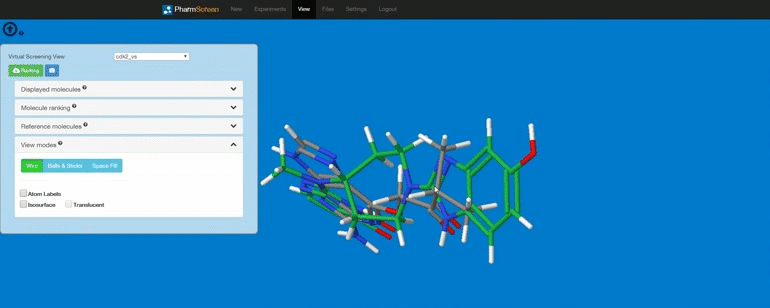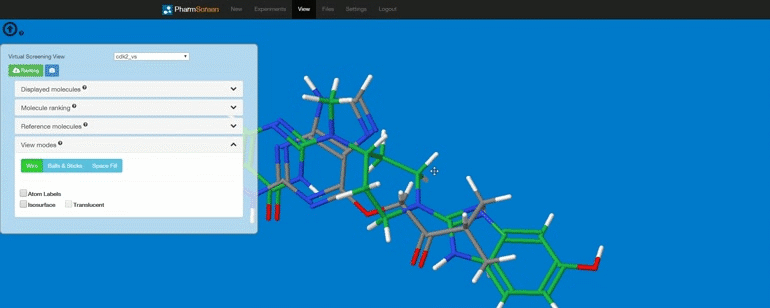
Requirements
What are the computational requirements to use PharmScreen GUI? Mainly none! You just need a web browser on your laptop/desktop.
[button text=”Ask for a demo!” link=”https://pharmacelera.com/ask-for-a-demo/” style=”default” size=”normal” target=”_self” display=”inline” icon=”no”]