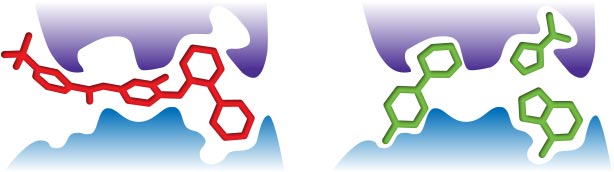
Image obtained from Advances in Fragment-Based Drug Discovery by Laura Elizabeth Mason
Fragment-Based Drug Design (FBDD) is becoming a useful strategy for hit to lead optimization in drug discovery. In the last years, some compounds derived from such methodology have progressed into clinical trials and it is estimated that this trend will increase in a near future . Some successful examples are HSP90 inhibitors developed by Abbot, protein kinase B (PKB) inhibitors by Astex and FDA approved drugs like Vemurafenib and Venetoclax.(1)
FBDD is based on the identification and selection of fragments which bind to sub-pockets of the target protein (2). Fragments are small molecules with attractive properties such as low molecular weight, and less chemical complexity and higher polarity and solubility than larger molecules. These identified fragments are evolved into more complex compounds by linking, growing or merging together the fragments. The result of this optimisation process leads into compounds with better pharmacokinetics, physical and toxicology properties (3).
FBDD shows clear advantages when compared with conventional High-Throughput Screening (HTS) (1,2).
- Fragments are less complex than larger molecules. This is translated in better fitting into specific cavities of the target binding site
- FBDD covers a greater chemical space in comparison with conventional libraries
- Hit fragments lead to higher chemical diversity than conventional HTS
- The optimization process is more successful, normally reducing the time of drug discovery
Fragment-Based Drug Discovery process
The first step involves the design of a tailored fragment library. The quality of the same will affect the final results of the whole FBDD. Subsequently, several restrictions and criteria are applied to the molecule fragments selected when building a specific library. Chemoinformatics approaches play an important role during this phase to select those molecules more suitable for the screening (e.g. excluding reactive molecule and toxic molecules or looking for chemical diversity) (1). Target-focused libraries can also be generated by including pharmacophore screening.
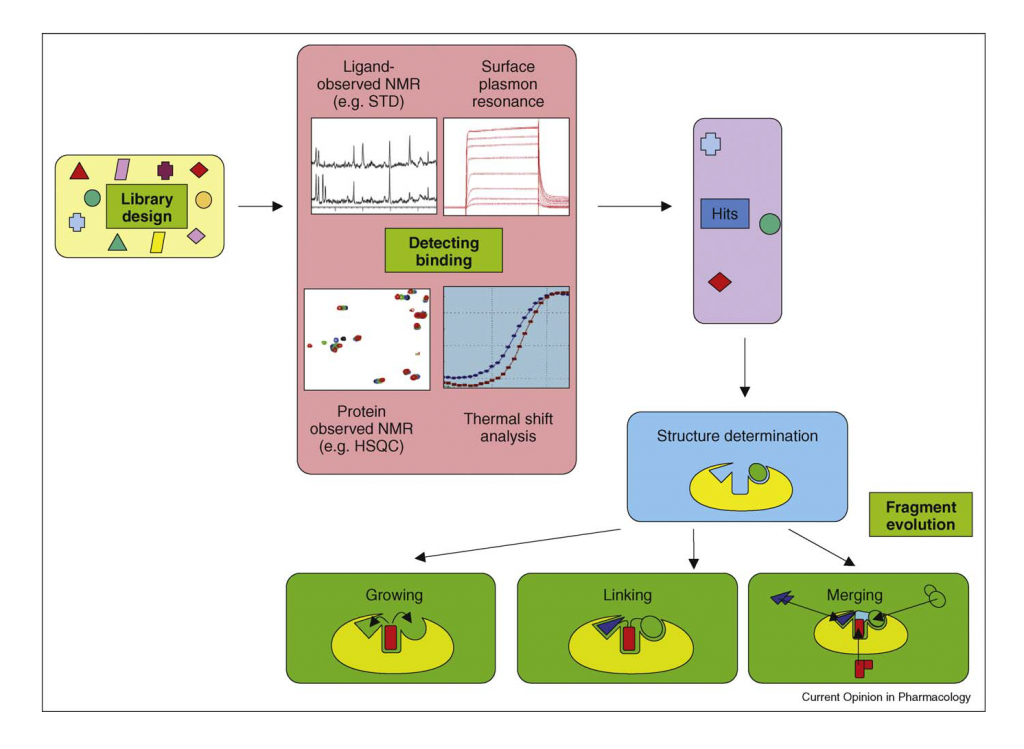
Essential steps in a FBDD project. Image obtained fromMichèle N Schulz and Roderick E Hubbard “Recent progress in fragment-based lead discovery”
Once the library is built or selected, the next step is to screen these fragments and check whether they bind to some sub-pocket in the target protein. Due to the low binding affinity of fragments, traditional biochemical assays are not suitable for hit identification in FBDD and biophysical approaches such as NMR, X-ray crystallography, isothermal titration calorimetry or plasmon resonance are employed. (1) (2)
Later, the identified active fragments are optimized and transformed into leads by (1) (3) (5):
- Fragment linking: Involves the linking of non-overlapping fragment which binds in adjacent sites of the target.
- Fragment merging: Overlapping hit fragment are merged by combining common structural parts
- Fragment growing: Using the structural information of the target, the fragment hit growing into a lead like compound is performed.
Field-Based computational approaches and FBDD
Pharmacelera’s technology uses a unique 3D representation of molecules based on electrostatic, steric and hydrophobic interaction fields derived from semi-empirical Quantum-Mechanics (QM) calculations. Such interaction field-based technology is especially synergistic with FBDD in the following aspects:
- False positive discrimination: A common difficulty when performing a FBDD project is the large amount of false positives founds during the HTS. The field-based representation of the molecules can be useful when identifying those fragments which are more likely to represent a positive hit, reducing an important waste of time and resources. (6)
- Screening the fragments: Field-based virtual screening can be used to virtually identify fragments that most likely bind to the target protein and that can be used for further optimization. The combination of steric, electrostatic and hydrophobic interaction fields, which are the most important factors that determine ligand-receptor interactions, identifying more active fragments than traditional methodologies
- Optimizing hits into lead candidates: field-based virtual screening technology can be applied when looking for similar compounds to the hit fragment in molecule databases. Our unique 3D representation of molecules allows finding molecules with similar physicochemical properties to reference fragment hit compound but with entirely different molecular scaffolds.
Are you working on FBDD? Do you want to know more about how our technology can help you increase your R+D productivity in this field? Contact us for an open discussion with no commitment!
(1) Michèle N Schulz1 and Roderick E Hubbard “Recent progress in fragment-based lead discovery”, Current Opinion in Pharmacology 2009, 9:615–621
(2) Kumar, Ashutosh & Voet, Arnout & Zhang, Kam. (2012). Fragment Based Drug Design: From Experimental to Computational Approaches. Current medicinal chemistry. 19. 10.2174/092986712803530467.
(3) Diane Joseph-McCarthy, Arthur J. Campbell, Gunther Kern, and Demetri Moustakas, “Fragment-Based Lead Discovery and Design”, J. Chem. Inf. Model. 2014, 54, 693−704
(4) David C. Rees, Miles Congreve, Christopher W. Murray and Robin Carr, “FRAGMENT-BASED LEAD DISCOVERY”, Nature Reviews Drug Discovery 3(8):660-72 · September 2004
(5) Laura Elizabeth Mason “Advances in Fragment-Based Drug Discovery” Technology Networks
(6) A False-Positive Screening Hit in Fragment-Based Lead Discovery: Watch out for the Red Herring. Angewandte Chemie (International ed. in English). 56. 10.1002/anie.201609824.